

Key Questions
Key scientific questions we are addressing include:
(i) How is the NRF2 signaling network regulated in response to diverse environmental cues?
(ii) How does a prolonged NRF2 response (dark side) during chronic arsenic exposure lead to lung cancer and type II diabetes?
(iii) How can we rationally design therapeutics targeting NRF2?
(iv) What are the overlapping/distinct regulatory mechanisms/roles of NRF1, NRF2 and NRF3?
My laboratory will pursue answers to these questions through innovative, rigorous, and multitiered approaches designed to delineate NRF biology and generate NRF-based therapeutics to mitigate disease.
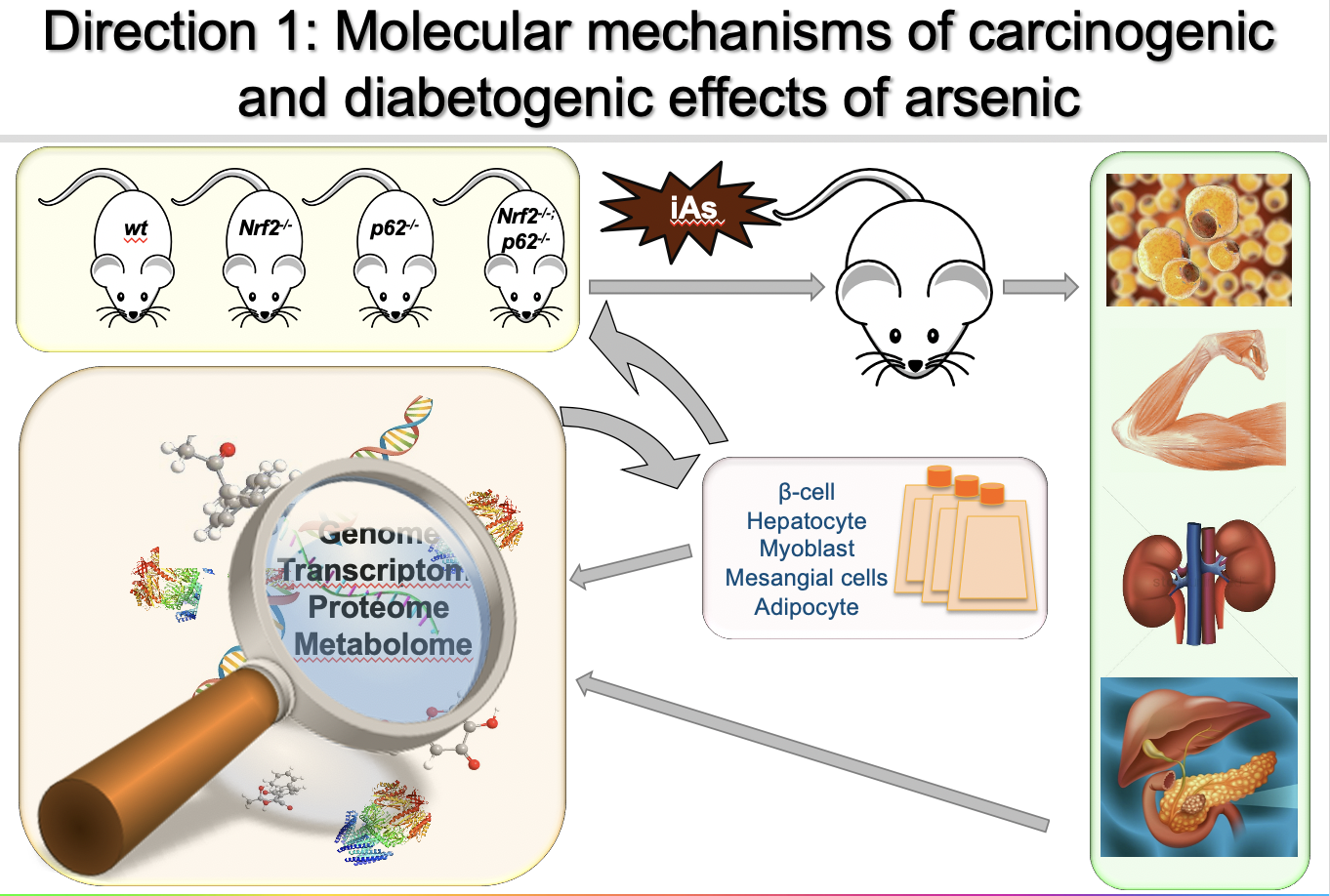
Direction 1: Molecular mechanisms of NRF2 dysregulation (“dark side”) that drive the carcinogenic and diabetogenic effects of arsenic
Arsenic-associated pathologies, including cancer and type II diabetes, affect millions of people worldwide; however, very little is known regarding the molecular mechanisms that drive these diseases. Based on our ongoing studies indicating that NRF2 dysregulation plays a critical role in these diseases, my long-term goal is to identify key NRF2 targets that drive the pathogenic effects associated with arsenic. To this end, my group has shown that arsenic blocks the autophagy-lysosome pathway, resulting in the p62-dependent activation of NRF2 (non-canonical mechanism). Although it is known that the intermittent oxidative or electrophilic activation of NRF2 (canonical mechanism) has protective effects against acute environmental insults, hyperactivation of NRF2 can result in a significant shift in the overall redox, metabolic, and proteostatic status of the cell. Thus, we believe that prolonged activation of the NRF2 signaling cascade due to arsenic-mediated autophagy inhibition (proteotoxic stress) could be a key factor in arsenic-associated pathogenesis. We are currently testing this hypothesis in two disease models, lung cancer and type II diabetes.
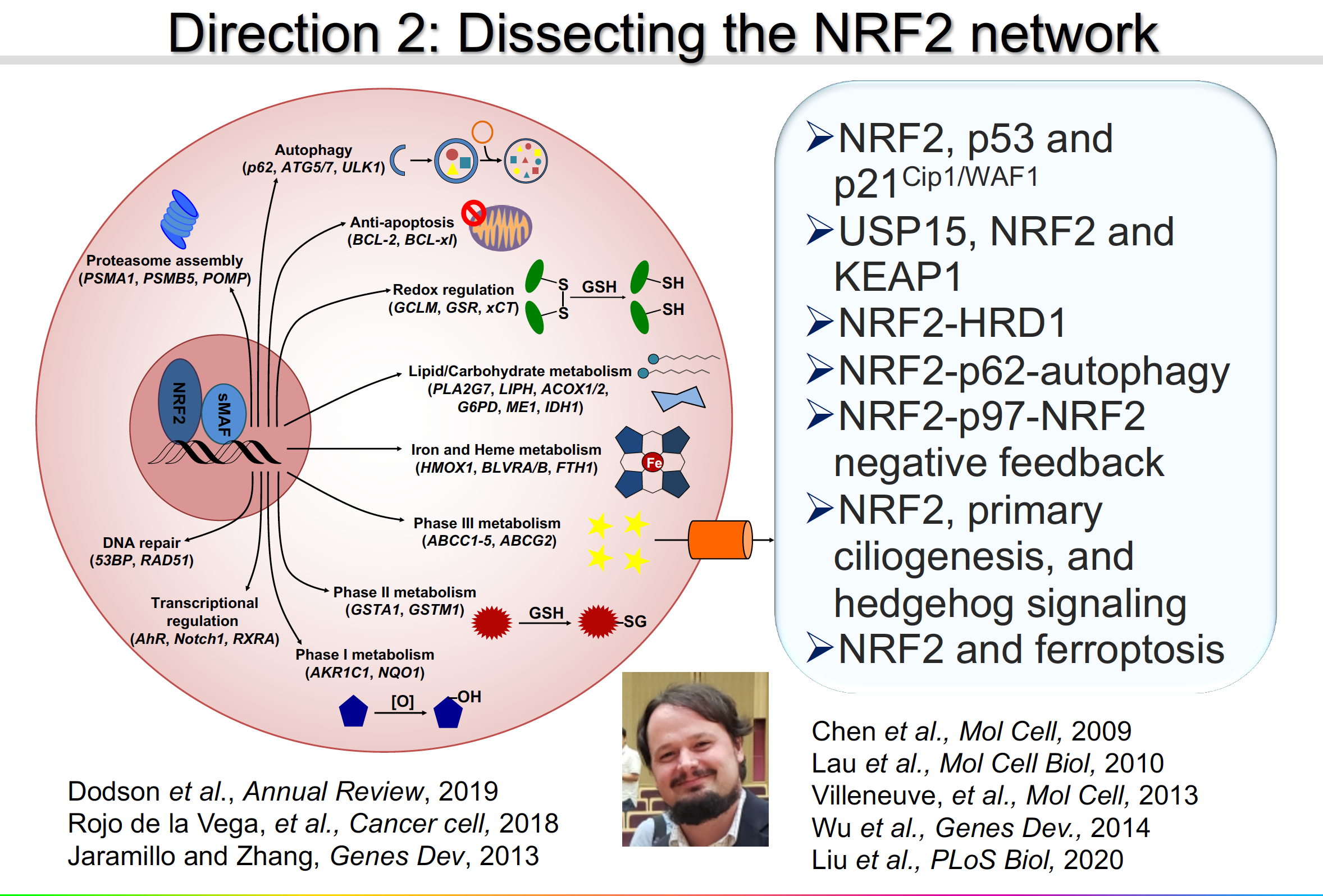
Direction 2: Dissecting the NRF2 signaling network
My laborataory is at the forefront of unveiling the molecular mechanisms of NRF2 regulation in response to diverse environmental cues/pathological states. We will continue to refine our understanding of the interconnected nature of the NRF2 signaling network. We are investigating cross-talk between the NRF2 pathway and other signaling pathways, as well as the pathophysiological roles of such cross-talk. The following are a few examples: NRF2-KEAP1-CUL3-RBX1; NRF2-KEAP1-p62-autophagy; NRF2-HRD1; NRF2-p53-p21Cip1/WAF1; NRF2-KEAP1-USP15; NRF2-p97-NRF2 negative feedback loop; NRF2,-primary ciliogenesis-hedgehog signaling; and NRF2-ferroptosis.
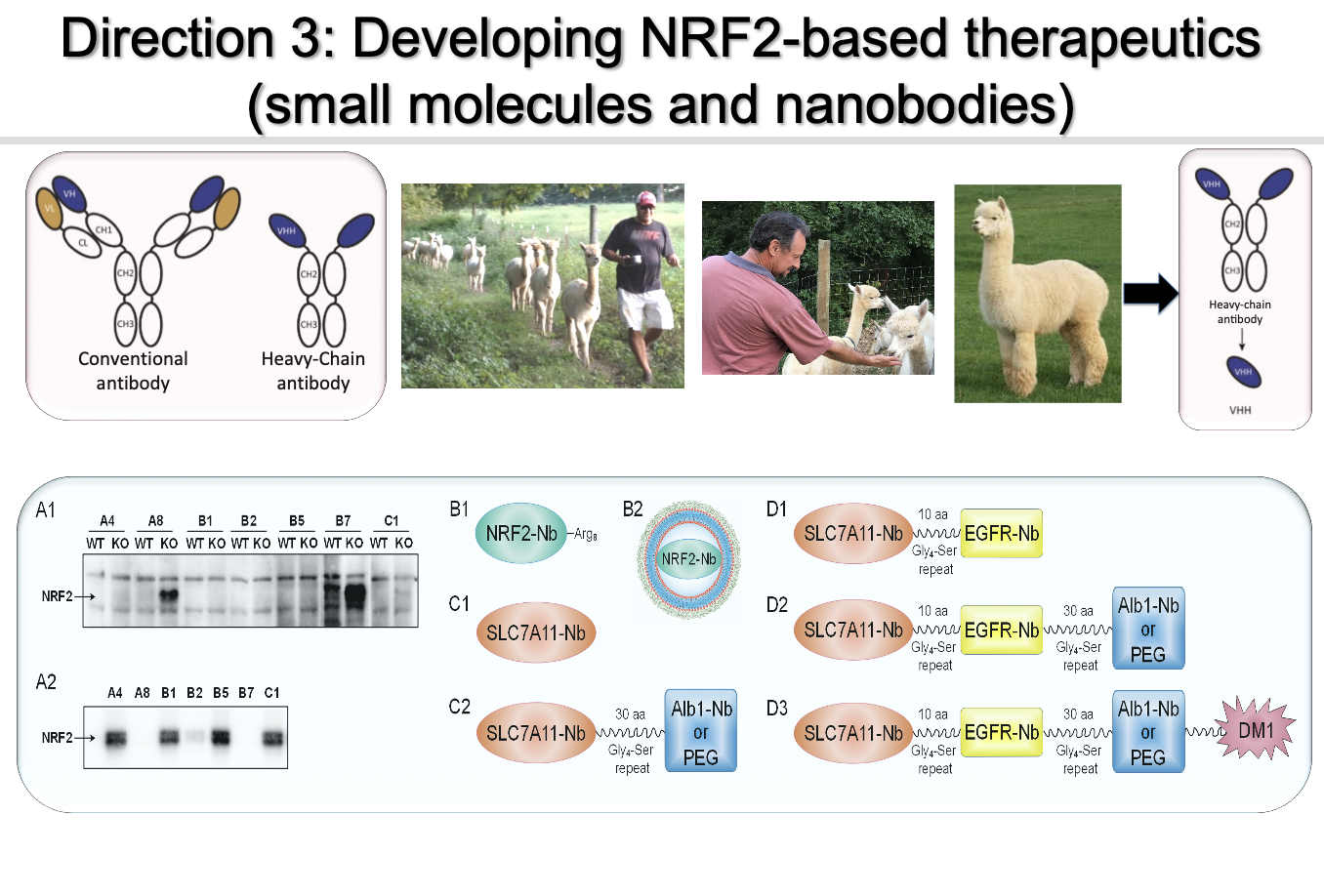
Direction 3: Development of probes/therapeutics targeting the “dark side” of NRF2
I envision that it is feasible to generate NRF2-based therapies based on the experimental knowledge my lab has accumulated over 20 years of NRF2 research. Somatic mutations in KEAP1 or NRF2, which result in constitutively elevated NRF2 protein levels, have been found in many cancer types including lung, head and neck, and bladder cancer. In fact, KEAP1 is mutated in lung cancer as frequently as the TP53 tumor suppressor gene (>30%). Cancer cells with constitutively high NRF2 levels rely on hyperactivation of the NRF2 response to survive, becoming “NRF2-addicted”. Clinically, high NRF2 expression in patient tumors is strongly correlated with a poor prognosis. Mounting evidence from my team and others convincingly indicates that uncontrolled NRF2 activation is a driver of cancer progression, metastasis, and resistance to therapy. Therefore, I hypothesize that NRF2-addicted cancers will be particularly vulnerable to NRF2 inhibition. We are developing small molecule NRF2 inhibitors and NRF2-based nanobodies as probes/therapeutics.
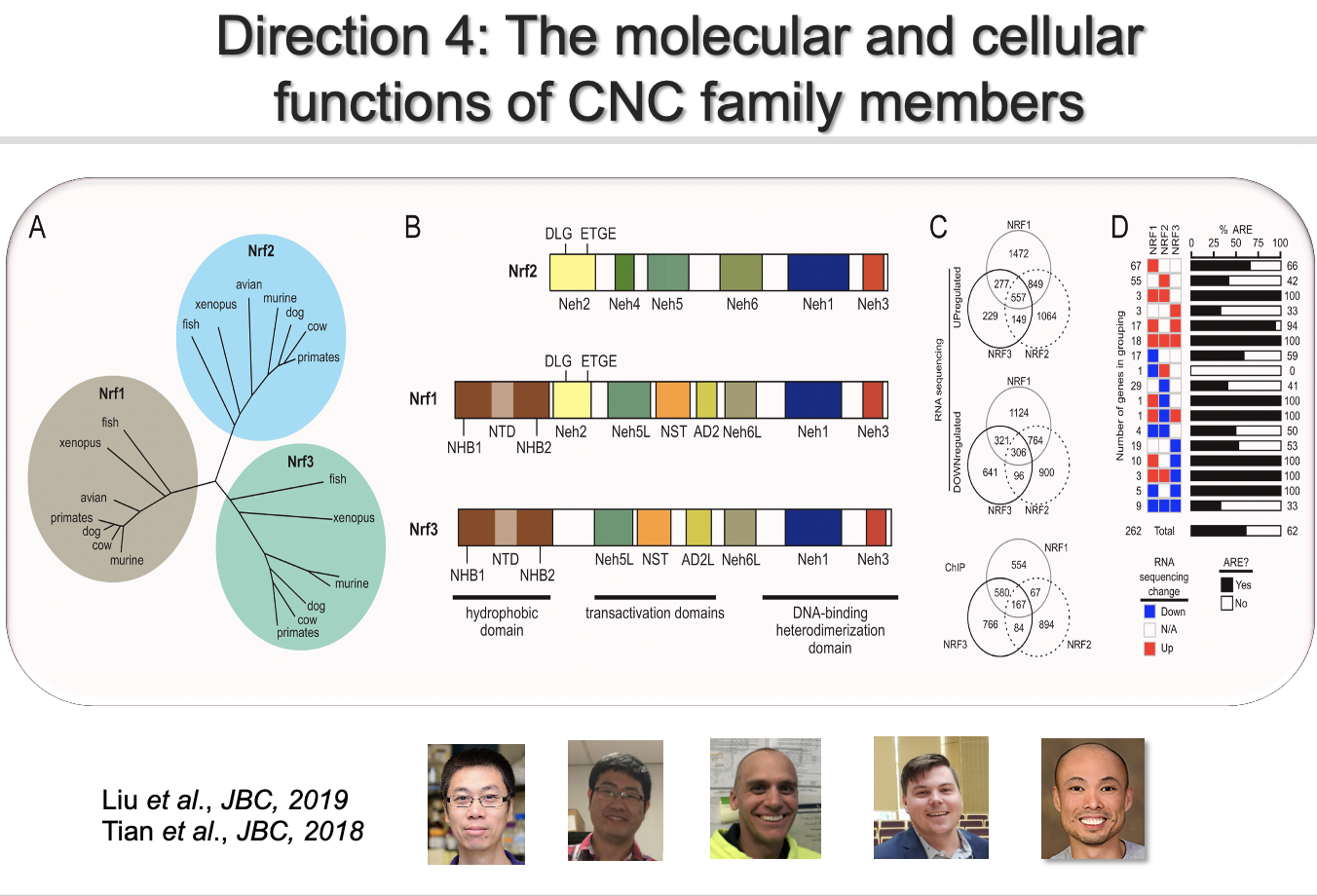
Direction 4: The molecular biology of the CNC family
NRF1, NRF2, and NRF3, belong to the CNC subfamily of the basic region leucine zipper transcription factors, are conserved across Euteleostomi, and homologues are found in lower organisms such as nematodes. NRF1, NRF2, and NRF3 may have distinct biological functions based on the observation that NRF1 knockout is embryonic lethal in mice, whereas NRF2 or NRF3 knockout mice are developmentally normal. The domain structures of NRF1, NRF2, and NRF3 also differ: NRF1 and NRF3 contain an extra N-terminal ER-associated sequence (Fig. 2B), which restricts them to the ER under basal conditions, whereas NRF2 is mainly localized in the cytoplasm. The evolutionary coexistence of all three NRF members indicates that each member has a unique function. However, the research in this area, specifically understanding the function of NRF1 or NRF3, is in its infancy. Several fundamental questions still need to be answered before we can truly understand/appreciate NRF biology. These key questions include the activation signals, the temporal and spatial response to a signal, and the set of downstream genes regulated by NRF1, NRF2, and NRF3. We are currently dissecting the molecular bases of the distinct and overlaping functions and phenotypes attributed to each NRF member.